Синдром Арнольда — Киар
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Аномалия Арнольда Киари
БЫСТРАЯ НАВИГАЦИЯ — Аномалия Арнольда Киари
Что такое Аномалия (Синдром) Арнольда Киари
Типы синдрома Арнольда-Киари
Согласно современной классификации выделяют четыре типа синдрома Арнольда Киари:
Последние два типа заболевания являются несовместимыми с жизнью.
Очень часто пациенты с синдромом Арнольда Киари имеют сопутствующую патологию позвоночника, которая возникает при сильном давлении анатомических структур задней черепной ямки на шейный отдел позвоночника. Такой патологией являются спинномозговые кисты. Они могут иметь большие размеры и часто вызывают симптоматику спинномозговых грыж (сильные боли, дисфункция внутренних органов).
Причины развития аномалии Арнольда Киари
Сегодня, врачи выделяют несколько причин, которые могут привести к развитию данного порока:
Симптомы и клиническая картина
Остальные пациенты имеют в той или иной степени выраженную симптоматику болезни. Заболевание сопровождается болевым синдромом в шейно-затылочной области головы, которая имеет свойство усиливается при кашле, икоте, а также при чихании. Врач-невропатолог находит снижение или полное отсутствие болевой, а также температурной чувствительности верхних конечностей, мышечную слабость и спастичность конечностей. Также, наблюдаются обмороки и головокружения, может быть снижения зрения, эпизодическое апноэ, непроизвольное быстрое движение глаз. Увеличение внутричерепного давления характеризуется утренней болью в голове, ощущением шипения и звона в ушах. Ухудшение координации проявляется тремором верхних или нижних конечностей. По мере прогрессирования болезни, у пациента появляются проблемы с мочеиспусканием, нарастают слабость и общая усталость. Во время движения клиническая картина стаёт более яркой.
Возможные осложнения
Неконтролируемое прогрессирование заболевание чревато появлением весомых осложнений. Они возникают не у всех больных, но порой всё же встречаются. К ним относится:
Диагностическая тактика
Если у пациента имеются определённые жалобы или аномалия развития была обнаружена случайно, то он должен обратиться к квалифицированному невропатологу для дальнейшей диагностики и лечения. Невропатолог проведёт сбор анамнеза, полный осмотр пациента и оценит его неврологический статус. Для постановки окончательного диагноза израильские специалисты используют следующие методы диагностики:
Аномалия Арнольда Киари — лечение в Израиле
Лечение каждого пациента зависит от ряда факторов. К ним относится тип аномального развития, длительность течения болезни, ступень тяжести, а также результаты всех диагностических процедур пациента и его индивидуальные особенности. На основании этих данных невропатолог составляет индивидуальное лечение.
В случае если основой клинической картины является незначительная головная боль, то можно вполне обойтись консервативным лечением с помощью противовоспалительных препаратов и миорелаксантов. А если заболевание прогрессирует и консервативная терапия неэффективна или малоэффективна, то нужно обратиться к оперативному хирургическому лечению.
В ходе операции нейрохирург ликвидирует сдавление нервных волокон и восстанавливает нормальную циркуляцию спинномозговой жидкости. Для этого ему необходимо увеличить заднюю черепную ямку. Если операция прошла успешно пациент ощущает себя намного лучше, дыхание, двигательные функции и чувствительность приходят в норму.
Специалисты израильских клиник подходят комплексно к лечению синдрома Арнольда-Киари, каждый пациент получает индивидуальную, наиболее подходящую схему лечения.
Профилактические меры
Профилактика появления синдрома заключается в бережном отношении беременной женщины к своему здоровью и здоровью малыша. Ей рекомендуется принимать здоровую пищу, отказаться от курения и приёма алкоголя. Рекомендациям лечащего врача нужно следовать в полном объёме, это, также, касается и приёма лекарственных средств. Всё это поможет свести вероятность возникновения порока развития у малыша к минимуму.
Что такое синдром арнольда киари
Информация о работе и расписание
Госпитальная высококвалифицированная медицинская помощь
Услуги центра по восстановительной медицине
Восстановление после спортивных травм
Современная диагностика – шанс предупредить болезнь
Он-лайн консультации для врачей по сложным практическим случаям
Трудоустройство в ФГАУ ЛРЦ
Стандарты и порядки оказания медицинской помощи
Проведение этической экспертизы клинических исследований, медицинских испытаний
Статьи и презентации
Мальформация Киари III типа заключается в смещении мозжечка и части ствола мозга с мозговыми оболочками в менингоцеле, расположенное в шейно-затылочной области.
Мальформации Арнольда–Киари II и III типов могут сопутствовать признаки дисплазии нервной системы: полимикрогирия, гетеротопия коры, гипоплазия подкорковых узлов, дисгенезия мозолистого тела, патология прозрачной перегородки, утолщение интерталамического соединения, beaking tectum (клювовидный tectum), часто отмечают наличие перегиба сильвиевого водопровода (55%), кисты отверстия Мажанди, гипоплазия серпа и намета мозжечка, hemivertebrae, низкое расположение каудального отдела спинного мозга на уровне LIV–V позвонков и ниже.
Этиология заболевания в настоящее время не ясна. Имеются данные, свидетельствующие о роли генетического фактора в этиологии этого синдрома. Эктопия миндалин мозжечка в затылочное отверстие была обнаружена у трех монозиготных близнецов. После первого описания мальформации Cleland в 1883 г. появилось несколько теорий. Теория, подтверждаемая исследованиями Misao Nishikawa и соавторов, заключается в том, что из-за парааксиальной дисплазии мезодермального листка или первичного повреждения структур соответствующего сомита формируется ненормально маленькая задняя черепная ямка, структуры заднего мозга, заполнив объем задней черепной ямки и продолжая расти, опускаются в затылочный канал. Сочетание Аномалии Киари II типа с менингомиелоцеле связано с тем, что степень парааксиальной дисплазии мезодермального листка при АК – II типа более выражена, чем при АК – I типа и отмечается не только на уровне формирования затылочной кости, но и по оси тела на уровне формирования ряда позвонков, что проявляется в spina bifida, а также в аномалиях ряда других костных структур и костной системы в целом.
Клинические проявления АК – I типа проявляются чаще всего в юношеском либо в зрелом возрасте. Эти проявления укладываются в такие неврологические синдромы, как церебеллобульбарный, ликворогипертензионный, сирингомиелический, синдромы повреждения черепных нервов. Ликворогипертензионный синдром проявляется головной болью, обычно субокципитальной, и болью в шее, усиливающейся при кашле, чихании и напряжении, застойными дисками зрительных нервов. Стволовые нарушения и расстройства функций черепных нервов проявляются в виде неустойчивых осциллопсий, тригеминальной дизестезии, снижения слуха, шума в ушах, головокружения, дисфагии, остановки дыхания во время сна, периодических обмороков (часто связанных с кашлем), нарушения контроля над ЧСС, АД при переходе из горизонтального положения в вертикальное, могут наблюдаться атрофия половины языка, паралич голосовых связок, стридор, спастический или комбинированный (больше в верхних конечностях) тетрапарез.
Мозжечковые расстройства — нистагм, дизартрия, атаксия. Симптомы, связанные с сирингомиелическими кистами — онемение, расстройство чувствительности, обычно по диссоциированному типу, а также нейроартропатия, нарушение функций тазовых органов, отсутствие брюшных рефлексов, мышечная гипотрофия. При этом ряд авторов отмечают несоответствие между локализацией, протяженностью кисты, кистозным индексом (отношение переднезаднего размера кисты к таковому размеру поперечника спинного мозга на уровне кисты), с одной стороны, и зоной гипестезии, распространенностью сегментарных расстройств поверхностной чувствительности, выраженностью мышечной гипотрофии и степенью пареза — с другой. АК II типа манифестирует у новорожденных и в раннем детском возрасте такими симптомами, как апноэ, стридор, билатеральный парез голосовых связок, нейрогенная дисфагия с назальной регургитацией, цианоз во время кормления, нистагм, гипотония, слабость, спастика в верхних конечностях, что может прогрессировать вплоть до тетраплегии. Мальформация Киари III типа встречается редко, клинические проявления ее такие же, как при АК II.
Стандартное рентгенологическое исследование может выявить лишь косвенные признаки мальформации АК, компьютерная томография также не дает четкой визуализации мягкотканных структур. Широкое внедрение МРТ в клиническую практику позволило решить большинство проблем, связанных с диагностикой аномалии Киари. Этому способствовали хорошая визуализация структур задней черепной ямки, краниовертебрального перехода, спинного мозга, отсутствие артефактов от костных структур.
Ориентиры задней черепной ямки, используемые в диагностике АК. d + e = длина ската; S = сфеноокципитальный синхондроз; d = длина основания сфеноидальной площадки от спинки турецкого седла и сфеноокципитального синхондроза до ската; e = длина между синхондрозом и basion; b = длина ствола мозга между плоскостью соединения среднего мозга и моста и медулло-цервикальным соединением; a = угол намета мозжечка по отношению к линии Твайнинга (Twining’s line); c = длина полушария мозжечка; DS = верхушка спинки турецкого седла; IOP = внутреннее возвышение затылочной кости; OP = opisthion; B = basion; TW = линия Твайнинга; McR (B to OP) = линия МакРи (McRae’s line). (заимствовано из Dimensions of the posterior fossa in patients symptomatic for Chiari I malformation but without cerebellar tonsillar descent, Raymond F Sekula и соавт.).
Аномалия Арнольда-Киари – диагностика и лечение

Аномалия Арнольда-Киари – это неправильное анатомическое соотношение костных структур верхней части позвоночника, затылочной части и основания черепа с нижней частью головного мозга. В норме продолговатый мозг, варолиев мост и мозжечок находятся внутри черепной коробки. А головной мозг плавно трансформируется в спинной на уровне большого затылочного отверстия. В нем нервные структуры, позвоночные артерии и ликворопроводящие пути располагаются свободно.
При мальформации Арнольда-Киари часть конечных отделов головного мозга выходит за пределы черепа и спускается в отверстие черепа. В итоге сдавливаются ткани головного, шейного отдела спинного мозга, позвоночные артерии и ликворопроводящие пути. Как правило, это врожденная патология, реже – следствие перенесенных в раннем возрасте черепно-мозговых травм, нарушивших правильное формирование костей задней черепной ямки. Аномалия нередко комбинируется с другими пороками – сирингомиелическим синдромом, незаращением дужек шейных позвонков. Распространенность клинически значимых и тяжелых форм составляет тысячные доли процента.
Клиническая картина зависит от типа аномалии и нарушений функции сдавливающихся структур. Течение колеблется от бессимптомного до несовместимого с жизнью, в зависимости от типа.
Виды мальформации Киари и симптомы
Первый тип наиболее распространен. За пределы черепа выходят только миндалины мозжечка. Симптоматика часто отсутствует или выражена незначительно.
Второй тип характеризуется смещением в позвоночный канал почти всего мозжечка и продолговатого мозга. Как правило, сопровождается выраженными и многообразными клиническими проявлениями. Могут отмечаться синдромы:
Третий и четвертый типы приводят к смерти в первые дни жизни из-за тяжелейшей компрессии жизненно важных нервных структур, множественных аномалий области перехода черепа и позвоночника, недоразвития мозжечка.
Диагностика при аномалии Киари
Многообразие клинических проявлений и их неспецифичность не позволяют абсолютно достоверно связать нарушения с аномалией Арнольда-Киари до выполнения магнитно-резонансной томографии (МРТ). Только это исследование позволяет:
Детей для выполнения МРТ погружают в медикаментозный сон, чтобы обеспечить их полную неподвижность.
Другие диагностические исследования (неврологический осмотр, офтальмоскопия, цветное дуплексное сканирование сосудов головы и шеи) необходимы для определения степени функциональных расстройств и планирования лечения.
Пациенты с мальформацией Арнольда-Киари первого типа без клинических проявлений нуждаются только в динамическом наблюдении для своевременного обнаружения симптомов и начала терапии. Нередко это становится необходимым лишь в зрелом возрасте, на четвертом-пятом десятилетии жизни.
Медикаментозная терапия при аномалии Арнольда-Киари
Иногда стабилизировать состояние позволяет медикаментозная терапия, с помощью которой можно:
Однако консервативная терапия не устраняет истинной причины нарушений. И если они выражены, без промедления переходят к хирургическому лечению, так как длительная компрессия структур мозга приводит к необратимым изменениям в них, не исчезающим даже после устранения сдавливания.
Хирургическое лечение аномалии Арнольда-Киари
Хирургическая коррекция аномалии Арнольда-Киари направлена на расширение костного канала, сдавливающего мягкие образования и устранение ликвородинамических нарушений. Сложность ее состоит в трудной доступности операционного поля, малой величине и высокой плотности расположения жизненно важных нервных центров и проводящих путей.
Операции у детей еще более проблематичны в силу малых размеров анатомических образований и незрелости систем организма. Поэтому они выполняются в условиях нейрохирургических стационаров, оснащенных самой современной операционной техникой и оборудованием для реабилитации послеоперационных больных. С детьми работают высококвалифицированные нейрохирурги, имеющие большой опыт подобных операций.
Современная нейрохирургическая тактика в отношении мальформации Арнольда-Киари предполагает выполнение операций краниовертебральной декомпрессии и шунтирования ликворопроводящей системы. В крупных клиниках развитых стран такие операции выполняются в сопровождении нейронавигации, с применением эндоскопической и микрохирургической аппаратуры. Это позволяет уменьшить тяжесть операционной травмы и предотвратить случайное повреждение важных областей.
Кроме первичных, нейрохирургам ведущих зарубежных центров нередко приходится выполнять ревизионные операции маленьким иностранным пациентам после неудачных вмешательств, выполненных у них на родине. Проводится:
В ходе операции вскрывается и твердая мозговая оболочка. В разрез вшивают для облегчения циркуляции спинномозговой жидкости «заплату» из синтетического материала или собственных тканей пациента, например, фасции бедра.
Нейрохирургическая коррекция показана практически всем детям со вторым типом аномалии Арнольда – Киари. Своевременно выполненная операция позволяет предупредить необратимое повреждение мозговых структур и минимизировать функциональные нарушения почти в 90% случаев.
Мальформация Киари
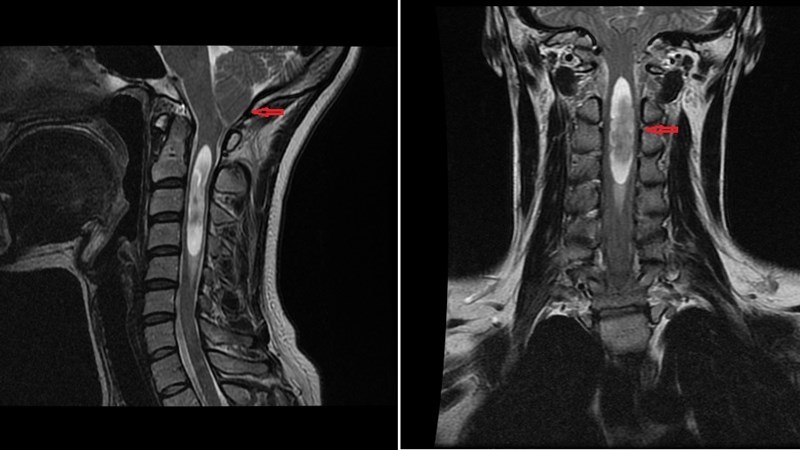
Мальформация Киари (термин Аномалия Арнольда-Киари правомочен для второго типа заболевания) является заболеванием, характеризующимся грыжевым выпячиванием миндаликов мозжечка в позвоночный канал через большое затылочное отверстие.
Признаки Мальформации Киари
Для пациентов взрослого возраста наиболее характерен первый тип Мальформации Киари, в то время как Мальформация Киари второго типа чаще проявляется и диагностируется у детей. В общем, при этом заболевании обнаруживается явное несоответствие в соотношении мозговых и костных структур на уровне краниовертебрального перехода. Происходит это за счет недоразвития костных элементов, формирующих, так называемую заднюю черепную ямку, где в норме уютно и комфортно расположен наш мозжечок. В результате этого на фоне нормально развивающихся мозговых структур этой области, миндалики мозжечка «выдавливаются» в позвоночный канал, меняют свою форму (становятся заостренными), сдавливают невральные и сосудистые структуры и проводящие пути в этой зоне и приводят к нарушению нормального процесса ликвороциркуляции – оттока спинномозговой жидкости.
Именно из-за этого наиболее частыми проявлением Мальформации Киари является, так называемая кашлевая головная боль. То есть головная боль в шейно-затылочной области, усиливающаяся при кашле, натуживании, игре на духовых инструментах и т.п.
К другим, наиболее часто обнаруживающимся симптомам Мальформации Киари относится дискоординация (шаткость при ходьбе, неловкость при выполнении точных движений, изменение почерка и т.п.), осиплость голоса, поперхивание при еде, онемение и слабость в конечностях.
При наличии МР признаков Мальформации Киари целесообразно выполнение исследования шейного отдела позвоночника для исключения/подтверждения сопутствующей сирингомиелии.
Лечение Мальформации Киари
В настоящее время известно более 20 видов хирургического вмешательства при Мальформации Киари. Все они, так или иначе, направлены на устранение несоответствия соотношения костных и нервальных структур. Диапазон варьирует от устаревших радикальных методик, сопровождающихся резекцией (удалением) миндаликов мозжечка, до малоинвазивных методик ограниченных резекцией костных элементов уровня краниовертебрального перехода, наиболее часто применяемой у детей.
При наличии сопутствующей гидроцефалии, в качестве первого этапа хирургического лечения необходимо выполнение эндоскопической вентрикулостомии дна третьего желудочка, практически полностью вытеснившей устанавливаемые ранее силиконовые шунтирующей системы. У определенного процента пациентов, страдающих от гипертензионно-гидроцефального синдрома, бывает достаточно лишь выполнения ЭТВ для купирования имеющихся жалоб. Однако большинству пациентов, страдающих именно от симптомов поражения структур задней черепной ямки и краниовертебрального перехода, требуется выполнение краниовертебральной декомпрессии.
Общепринятым золотым стандартом хирургической коррекции Мальформации Киари I типа, является операция – декомпрессии уровня краниовертебрального перехода с пластикой твердой мозговой оболочки. Суть метода заключается в устранении существующей компрессии на структуры этой области, увеличении «полезного» объема задней черепной ямки и нормализации процесса ликвороциркуляции. Следует отметить, что несмотря на то что положительные результаты при достаточном опыте хирурга и адекватно выполненном оперативном вмешательстве могут быть достигнуты более чем в 80% случаев, задачей хирурга является, как минимум, остановить патологический процесс. То есть не допустить дальнейшего нарастания неврологической симптоматики у пациентов с Мальформацией Киари.
Быстрее всего в послеоперационном периоде регрессируют характерные для большинства пациентов упорные головные боли и нарушение координации. Регресс симптомов, связанных непосредственно с формированием сирингомиелии, наступает чуть позже. Для оценки результатов хирургического лечения в среднем через шесть месяцев после операции выполняется контрольное МРТ исследование, позволяющее судить о нормализации соотношения структур на уровне краниовертебрального перехода, динамике размеров сирингомиелической полости. Однако, при оценке результатов хирургического лечения, в первую очередь, мы ориентируемся именно на клиническую картину, то есть на динамику симптомов заболевания, как наиболее важную составляющую для каждого нашего пациента.